
This is one of the most common questions that we hear from almost all the teams we met during our Regulatory Compliance Clinic. Most people would prefer not to get their product into the medical device space, while some prefer to get their products into this regulated space to differentiate themselves from competitors. Is this coming down to a choice or preference? Or is there a clear boundary to it? Let’s discuss!
Market Analysis

All the development teams we met so far have very interesting platform technologies. The team can readily translate them into very useful hardware or software applications. However, before we look at how great a product it can be, all teams must start thinking about the following – Where is the demand coming from? What pain points are you solving? Which markets are you serving? These are the many other customer-related questions in which programmes like the NUS Lean Launchpad are encouraging teams to uncover. This should bring you closer to crafting an “Intended Use” statement for your product, which is a market driven process.
Product Intended Use Statement
With an intended use statement, we are one step closer to identifying if your product is classified as a medical device.
From the Singapore HSA website, the definition a medical device is:
“Medical device” means any instrument, apparatus, implement, machine, appliance, implant, in vitro reagent or calibrator, software, material or other similar or related article that is intended by its manufacturer to be used, whether alone or in combination, for humans for one or more of the specific purposes of
(a) diagnosis, prevention, monitoring, treatment or alleviation of any disease;
(b) diagnosis, monitoring, treatment, alleviation of or compensation for an injury;
(c) investigation, replacement, modification, or support of the anatomy or of a physiological process;
(d) supporting or sustaining life;
(e) control of conception;
(f) disinfection of medical devices; or
(g) providing information for medical or diagnostic purposes by means of in vitro examination of specimens derived from the human body,
and which does not achieve its primary intended action in or on the human body by pharmacological, immunological or metabolic means, but which may be assisted in its intended function by such means.
All companies would need to note that this definition is quite harmonised across the various countries, although there might be slight differences which may affect your product to be classified as a medical device or not.
For further understanding on ways to create your intended use statement, read on!
Medical Device Classification

Once you have determined the “Intended Use” and it falls within the medical device definition, the next step would be to understand which risk classification it falls into. Bear in mind that your product classification could vary in different countries because of the difference in the regulations. This is even more possible if you are developing a new and innovative medical device. E.g., A software might be classified as a Class B product in Singapore, but as a Class IIb in Europe.
Thus, it is important to first define the primary markets of entry, iron out the risk classification, understand the submission requirements and clearly list them out in the early stages of your product development. Addition of new markets unnecessarily may slow down the product development process, especially if engineering changes or additional tests need to be performed.
A Case Study
Scenario: A company would like to identify the risk classification of its new electrocardiogram (ECG) under Singapore HSA’s regulation.
Intended use – The Alpha ECG is intended to acquire, analyse, display and record ECG signals from electrodes by trained operators in a hospital or equivalent facility. The Alpha ECG is not designed to provide alarms for arrhythmia detection.
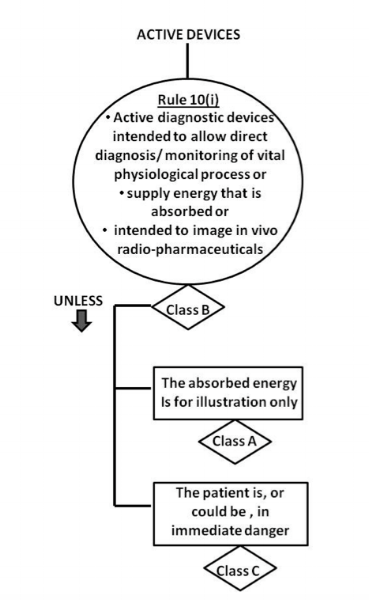
Since an ECG requires a power source to operate, it is generally known as an “active” medical device. This helps us to narrow down the options to between Rule 9 and Rule 12 of GN-13 Guidance on the Risk Classification of General Medical Devices. As stated in the intended use statement, the ECG information is acquired, analysed and recorded on a display. In an actual use case, this data aids the physician in his/her diagnosis of the patient’s cardiovascular status. In this instance, it would rightfully be classified under Rule 10(i) as a Class B device, where it is intended to diagnose/monitor the physiological condition of the heart. It is not a Rule 10(i) Class A, which is a device used for illumination (e.g., lighting) nor a Class C device used to diagnose patient in an immediate danger.
Using the intended use statement, this is how you can start out to identify the most correct classification for your medical device. Moving forward, you can also verify your identification with the result from the HSA risk classification tool to give yourself a better sense on steps to take.
(Note: the above case study is done based on an ECG classified as a general medical device. If you are dealing with an in vitro diagnostic (IVD) product, do refer to GN-14 Guidance on the Risk Classification of In Vitro Diagnostic Medical Devices for the classification rules.)
Determining the path to market

After knowing the classification of your medical device, you are now ready to determine the path needed to get your product registered with HSA. For more information and guidance regarding product registration, please refer to the following document – GN-15 Guidance On Medical Device Product Registration.
For companies in the product development stages, it is important to identify your intended use, risk classification and know what data/information you would need to prepare for regulatory submission.
Once done, start your development phase right with all the above considerations in mind!



